
1st Department of Pathology and Experimental Cancer Research

Contributions
Type: Oral Presentation + travel grant
Presentation during EHA20: From 12.06.2015 12:15 to 12.06.2015 12:30
Location: Room C2
Background
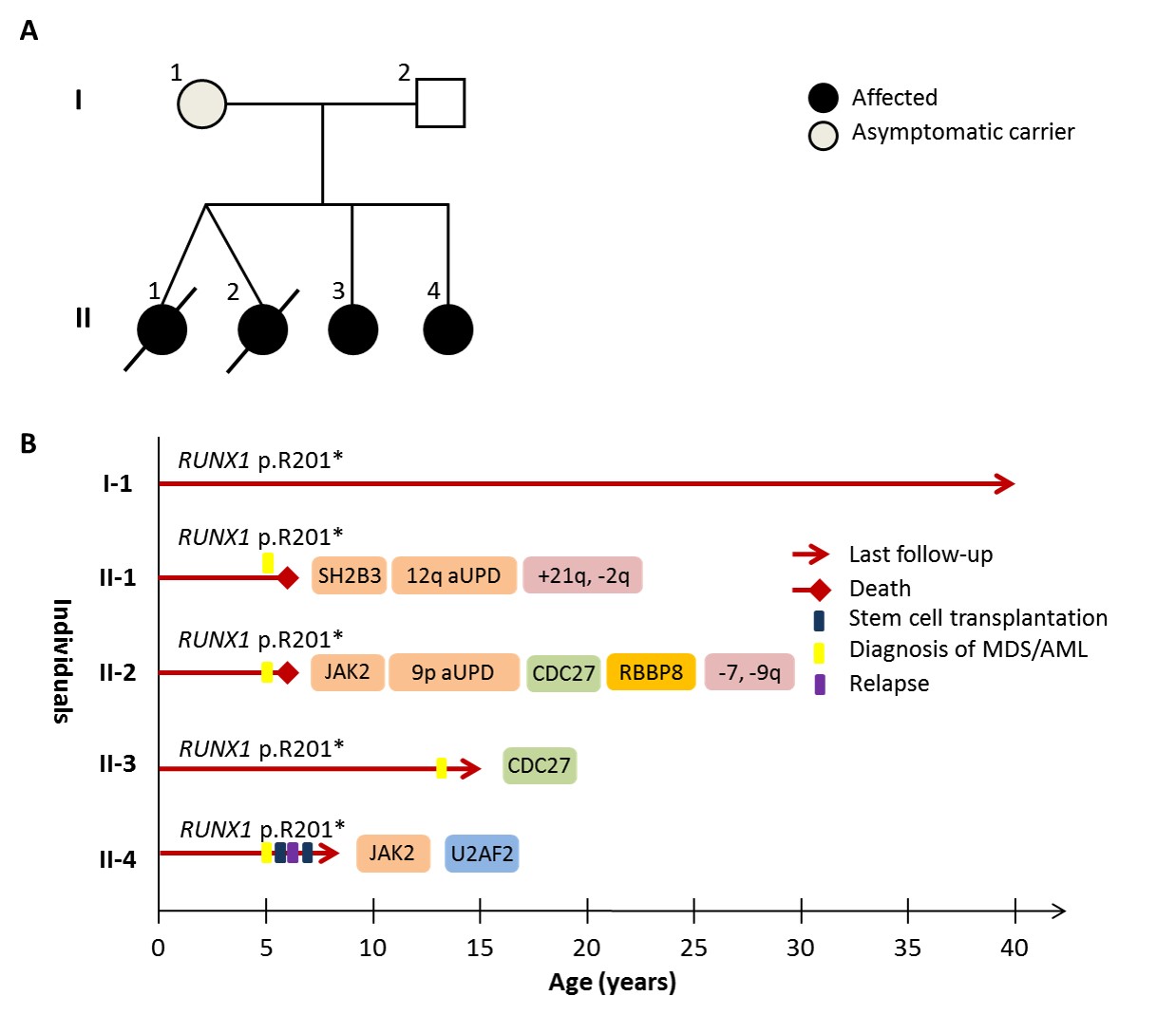
Germline mutations in the transcription factor RUNX1 confer an autosomal dominant predisposition to familial platelet disorder (FPD) and myelodysplasia/acute myeloid leukaemia (MDS/AML). The penetrance of mutations varies with approximately 40% of carriers developing overt malignancy, whilst others remain asymptomatic or manifest mild to moderate FPD. We describe a novel RUNX1-mutated family, where 3 young siblings presented with secondary AML, providing a rare opportunity to compare the molecular events initiating disease (Figure 1A).
Aims
To genetically profile somatic aberrations in multiple cases of MDS/AML from a novel FPD/AML pedigree
Methods
We performed whole exome sequencing (WES) on bone marrow (BM) DNA from 4 siblings with MDS/AML with an average exonic coverage of 96x. Peripheral blood (PB) DNA samples from both healthy parents were also sequenced to enable exclusion of inherited variants in the 4 children. Acquired mutations, copy number aberrations (CNA) and loss of heterozygosity (LOH) were then defined across the four siblings. Key mutations were confirmed with Sanger sequencing, whilst further verification of CNA was performed using multiplex ligation-dependent probe amplification (MLPA).
Results
Direct Sanger sequencing of the 4 siblings (II.1-II.4) and their mother (I.1) revealed the germline RUNX1 mutation, p.R201X. Figure 1B summarises the clinical timeline, with the key molecular and cytogenetic lesions detected in MDS/AML from each sibling. Their mother (45y) remains an asymptomatic carrier, with no peripheral cytopenias. The dizygotic twins (II.1 and II.2) presented within a period of 2 weeks at 5y, both with hepatosplenomegaly and pancytopenia. BM morphology of each twin revealed AML with dysplastic and myelomonocytic features. Significant somatic chromosomal aberrations included gain of 21q (II.1), monosomy 7 and deletion of chromosome 9q (II.2). Ten years later, sibling II.4 also presented at 5y with monocytosis. BM examination revealed myelomonocytic AML with dysplastic features and a normal cytogenetic profile. Sibling II.3 is now 14y, her BM examination revealed multi-lineage dysplasia and normal cytogenetics.
WES revealed molecular addiction to JAK2 signalling in 3 siblings (II.1, II.2 and II.4). II.2 and II.4 both acquired JAK2 V617F mutations, with homozygosity of the mutant allele observed in II.2 due to 9p acquired uniparental disomy (aUPD). In II.1, we detected a somatic mutation in SH2B3 (p.R392Q), with apparent homozygosity caused by aUPD of 12q. The p.R392Q mutation was localised to the SH2 domain, which normally binds both mutant and WT isoforms of JAK2, inhibiting their phosphorylation. Further somatic mutations occurred in CDC27 (anaphase promoting complex, II.2 and II.4), RBBP8 (DNA double-strand break repair, II.2) and U2AF2 (spliceosome complex, II.4). Notably, all 3 siblings with somatic JAK2-signalling lesions had aggressive disease. Both twins died within a year of presentation, II.1 due to relapse and II.2 from chemotherapy-refractory disease. Sibling II.4 relapsed after 13 months and is currently in CR2 following allogeneic HSCT.
Summary
We describe a novel FPD-AML pedigree demonstrating convergence of lesions within the JAK-STAT signalling pathway in 3 siblings with MDS/AML. Since JAK2 mutations are reported in <5% of sporadic RUNX1-mutated AML, our findings suggest somatic mutations in FPD/AML may be enriched within distinct signalling pathways, often associated with aUPD to increase the mutant allele burden within tumours.
Keyword(s): MDS/AML, RUNX1
Session topic: Molecular pathogenesis of AML
Type: Oral Presentation + travel grant
Presentation during EHA20: From 12.06.2015 12:15 to 12.06.2015 12:30
Location: Room C2
Background
Germline mutations in the transcription factor RUNX1 confer an autosomal dominant predisposition to familial platelet disorder (FPD) and myelodysplasia/acute myeloid leukaemia (MDS/AML). The penetrance of mutations varies with approximately 40% of carriers developing overt malignancy, whilst others remain asymptomatic or manifest mild to moderate FPD. We describe a novel RUNX1-mutated family, where 3 young siblings presented with secondary AML, providing a rare opportunity to compare the molecular events initiating disease (Figure 1A).
Aims
To genetically profile somatic aberrations in multiple cases of MDS/AML from a novel FPD/AML pedigree
Methods
We performed whole exome sequencing (WES) on bone marrow (BM) DNA from 4 siblings with MDS/AML with an average exonic coverage of 96x. Peripheral blood (PB) DNA samples from both healthy parents were also sequenced to enable exclusion of inherited variants in the 4 children. Acquired mutations, copy number aberrations (CNA) and loss of heterozygosity (LOH) were then defined across the four siblings. Key mutations were confirmed with Sanger sequencing, whilst further verification of CNA was performed using multiplex ligation-dependent probe amplification (MLPA).
Results
Direct Sanger sequencing of the 4 siblings (II.1-II.4) and their mother (I.1) revealed the germline RUNX1 mutation, p.R201X. Figure 1B summarises the clinical timeline, with the key molecular and cytogenetic lesions detected in MDS/AML from each sibling. Their mother (45y) remains an asymptomatic carrier, with no peripheral cytopenias. The dizygotic twins (II.1 and II.2) presented within a period of 2 weeks at 5y, both with hepatosplenomegaly and pancytopenia. BM morphology of each twin revealed AML with dysplastic and myelomonocytic features. Significant somatic chromosomal aberrations included gain of 21q (II.1), monosomy 7 and deletion of chromosome 9q (II.2). Ten years later, sibling II.4 also presented at 5y with monocytosis. BM examination revealed myelomonocytic AML with dysplastic features and a normal cytogenetic profile. Sibling II.3 is now 14y, her BM examination revealed multi-lineage dysplasia and normal cytogenetics.
WES revealed molecular addiction to JAK2 signalling in 3 siblings (II.1, II.2 and II.4). II.2 and II.4 both acquired JAK2 V617F mutations, with homozygosity of the mutant allele observed in II.2 due to 9p acquired uniparental disomy (aUPD). In II.1, we detected a somatic mutation in SH2B3 (p.R392Q), with apparent homozygosity caused by aUPD of 12q. The p.R392Q mutation was localised to the SH2 domain, which normally binds both mutant and WT isoforms of JAK2, inhibiting their phosphorylation. Further somatic mutations occurred in CDC27 (anaphase promoting complex, II.2 and II.4), RBBP8 (DNA double-strand break repair, II.2) and U2AF2 (spliceosome complex, II.4). Notably, all 3 siblings with somatic JAK2-signalling lesions had aggressive disease. Both twins died within a year of presentation, II.1 due to relapse and II.2 from chemotherapy-refractory disease. Sibling II.4 relapsed after 13 months and is currently in CR2 following allogeneic HSCT.
Summary
We describe a novel FPD-AML pedigree demonstrating convergence of lesions within the JAK-STAT signalling pathway in 3 siblings with MDS/AML. Since JAK2 mutations are reported in <5% of sporadic RUNX1-mutated AML, our findings suggest somatic mutations in FPD/AML may be enriched within distinct signalling pathways, often associated with aUPD to increase the mutant allele burden within tumours.
Keyword(s): MDS/AML, RUNX1
Session topic: Molecular pathogenesis of AML