

Contributions
Type: Oral Presentation
Presentation during EHA20: From 13.06.2015 11:30 to 13.06.2015 11:45
Location: Room Stolz 2
Background
In patients with hemoglobinopathies, hematopoietic stem cell (HSC) gene therapy has the potential to induce production of functional β-globin in the red blood cell lineage with the aim of reducing or eliminating the symptoms of disease. Results for 2 subjects with β0/βE-thalassemia major treated in clinical study HGB-205 that were previously presented (EHA 2014, ASH 2014) suggested that transplantation with autologous CD34+ cells transduced with the self-inactivating LentiGlobin BB305 lentiviral vector containing an engineered βA-T87Q?globin gene resulted in near-normal levels of total hemoglobin (Hb) and early transfusion independence.
Aims
Herein, we provide additional follow-up data on these two subjects as well as early safety and efficacy data on the first subject with sickle cell disease (SCD) treated with gene therapy.
Methods
After obtaining informed consent, subjects with β-thalassemia major undergo HSC collection via peripheral blood apheresis, while subjects with severe sickle cell disease undergo HSC collection via bone marrow harvest. CD34+ cells were selected and transduced with LentiGlobin BB305 lentiviral vector. Estimation of the mean ex- vivo vector copy number (VCN) was obtained by quantitative PCR performed on pooled colony-forming progenitors. Subjects underwent myeloablation with intravenous busulfan, followed by infusion of transduced CD34+ cells. Subjects were monitored for overall safety including hematological engraftment, βA-T87Q?globin expression (by high performance liquid chromatography) and transfusion requirements. Integration site analysis (ISA, by linear amplification-mediated PCR and high-throughput sequencing on nucleated cells) and replication-competent lentivirus (RCL) assays were performed. Prophylactic pRBC transfusions are continued in sickle cell subjects who are transfusion dependent pre-treatment to maintain HbS <30%, followed by gradual tapering over time.
Results
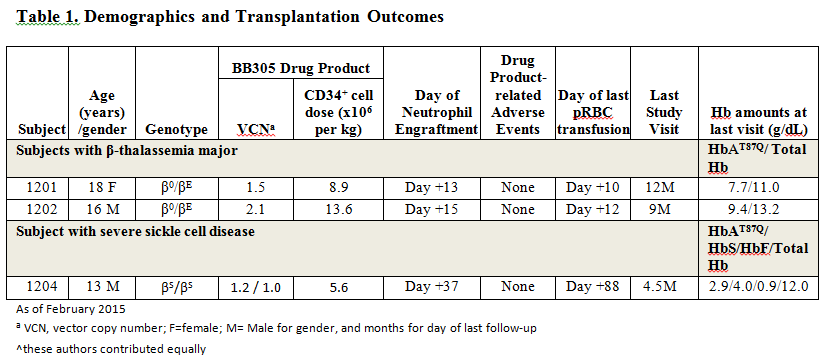
As of February 2015, 2 subjects with β0/βE thalassemia major (Subjects 1201 and 1202) and 1 subject with βS/βS severe sickle cell disease (Subject 1204, who was on prophylactic transfusion therapy for multiple veno-occlusive crises and acute chest syndrome) have undergone treatment. The outcome of these subjects to date is shown in Table 1. No subject has experienced a drug product related adverse event, and ISA analyses demonstrate highly polyclonal reconstitution without clonal dominance. Both β-thalassemia major subjects remain transfusion-free for 14 and 11 months respectively, post-treatment. Subject 1204, at his most recent visit (Month 4.5 post-treatment), has a total hemoglobin of 12.0 g/dl, of which 24% is HbAT87Q (from 9.6% at Month 3) and 33% is HbS (from 14.7% at Month 3), with increasing levels of transduced cells detected in peripheral blood (PBL VCN of 2.4 at Month 4.5). This subject has not had a post-treatment hospitalization for a SCD-related event and his transfusion support is being tapered.
Summary
The subjects with β-thalassemia major remain transfusion-free for 14 and 11 months. The subject with SCD demonstrates increasing production of HbAT87Q over time. Gene therapy using autologous HSC transduced with LentiGlobin BB305 lentiviral vector ex vivo is a promising approach for the treatment of patients with both β-thalassemia major and severe SCD.
Keyword(s): Beta thalassemia, Gene therapy, Lentiviral vector, Sickle cell disease
Session topic: Gene therapy, cellular immunotherapy and vaccination
Type: Oral Presentation
Presentation during EHA20: From 13.06.2015 11:30 to 13.06.2015 11:45
Location: Room Stolz 2
Background
In patients with hemoglobinopathies, hematopoietic stem cell (HSC) gene therapy has the potential to induce production of functional β-globin in the red blood cell lineage with the aim of reducing or eliminating the symptoms of disease. Results for 2 subjects with β0/βE-thalassemia major treated in clinical study HGB-205 that were previously presented (EHA 2014, ASH 2014) suggested that transplantation with autologous CD34+ cells transduced with the self-inactivating LentiGlobin BB305 lentiviral vector containing an engineered βA-T87Q?globin gene resulted in near-normal levels of total hemoglobin (Hb) and early transfusion independence.
Aims
Herein, we provide additional follow-up data on these two subjects as well as early safety and efficacy data on the first subject with sickle cell disease (SCD) treated with gene therapy.
Methods
After obtaining informed consent, subjects with β-thalassemia major undergo HSC collection via peripheral blood apheresis, while subjects with severe sickle cell disease undergo HSC collection via bone marrow harvest. CD34+ cells were selected and transduced with LentiGlobin BB305 lentiviral vector. Estimation of the mean ex- vivo vector copy number (VCN) was obtained by quantitative PCR performed on pooled colony-forming progenitors. Subjects underwent myeloablation with intravenous busulfan, followed by infusion of transduced CD34+ cells. Subjects were monitored for overall safety including hematological engraftment, βA-T87Q?globin expression (by high performance liquid chromatography) and transfusion requirements. Integration site analysis (ISA, by linear amplification-mediated PCR and high-throughput sequencing on nucleated cells) and replication-competent lentivirus (RCL) assays were performed. Prophylactic pRBC transfusions are continued in sickle cell subjects who are transfusion dependent pre-treatment to maintain HbS <30%, followed by gradual tapering over time.
Results
As of February 2015, 2 subjects with β0/βE thalassemia major (Subjects 1201 and 1202) and 1 subject with βS/βS severe sickle cell disease (Subject 1204, who was on prophylactic transfusion therapy for multiple veno-occlusive crises and acute chest syndrome) have undergone treatment. The outcome of these subjects to date is shown in Table 1. No subject has experienced a drug product related adverse event, and ISA analyses demonstrate highly polyclonal reconstitution without clonal dominance. Both β-thalassemia major subjects remain transfusion-free for 14 and 11 months respectively, post-treatment. Subject 1204, at his most recent visit (Month 4.5 post-treatment), has a total hemoglobin of 12.0 g/dl, of which 24% is HbAT87Q (from 9.6% at Month 3) and 33% is HbS (from 14.7% at Month 3), with increasing levels of transduced cells detected in peripheral blood (PBL VCN of 2.4 at Month 4.5). This subject has not had a post-treatment hospitalization for a SCD-related event and his transfusion support is being tapered.
Summary
The subjects with β-thalassemia major remain transfusion-free for 14 and 11 months. The subject with SCD demonstrates increasing production of HbAT87Q over time. Gene therapy using autologous HSC transduced with LentiGlobin BB305 lentiviral vector ex vivo is a promising approach for the treatment of patients with both β-thalassemia major and severe SCD.
Keyword(s): Beta thalassemia, Gene therapy, Lentiviral vector, Sickle cell disease
Session topic: Gene therapy, cellular immunotherapy and vaccination