

Contributions
Type: Oral Presentation
Presentation during EHA20: From 13.06.2015 11:30 to 13.06.2015 11:45
Location: Room Stolz 1
Background
Impairment of PP2A activity by its negative regulators SET and cancerous inhibitor of PP2A (CIP2A) plays an important role in the pathogenesis and progression of chronic myeloid leukaemia (CML). CIP2A is associated with increased proliferation in several human malignancies and its over-expression can cause cellular transformation. High levels are an adverse prognostic indicator in many malignancies. In CML a high CIP2A protein level at diagnosis in imatinib treated patients may be a biomarker for subsequent blast crisis. However, its role in CML patients receiving second generation tyrosine kinase inhibitors (2G TKIs) is not known.
Aims
Our aim was to investigate if the superior clinical outcome seen in patients treated with 2G TKIs could be explained by modulation of the CIP2A pathway.
Methods
CIP2A, PP2A, PP2AY307, c-Myc and E2F1 proteins were assessed by FACS. CIP2A, c-Myc and E2F1 were depleted in K562 and CD34+ cells using siRNA.
Results
In mononuclear cells taken at diagnosis from 20 chronic phase CML patients, those with high CIP2A levels significantly decreased following dasatinib or nilotinib treatment (p=0.007 and p=0.001 respectively) and this was accompanied by a significant decrease in c-Myc, (p=0.002). Y307 phosphorylated (inactive) PP2A also decreased in dasatinib (p=0.03) and nilotinib (p=0.04) treated cells, thus increasing PP2A activity, but imatinib had no significant effect. These differences in TKI effects are not mediated by differential suppression of BCR-ABL activity (as assessed using the pCrKL assay).
E2F1 transcriptionally regulates both c-Myc and CIP2A and has previously been shown to be over-expressed in CML. E2F1 is high in patients with high diagnostic CIP2A protein levels compared to those with low CIP2A protein levels (p=0.04). Following 1 month of clinical 2G TKI treatment, E2F1 protein levels significantly decreased (p=0.01) and this was accompanied by a decrease in mRNA levels of its transcriptional target CIP2A. In sharp contrast, imatinib did not suppress E2F1, and thus CIP2A mRNA levels remain unchanged.
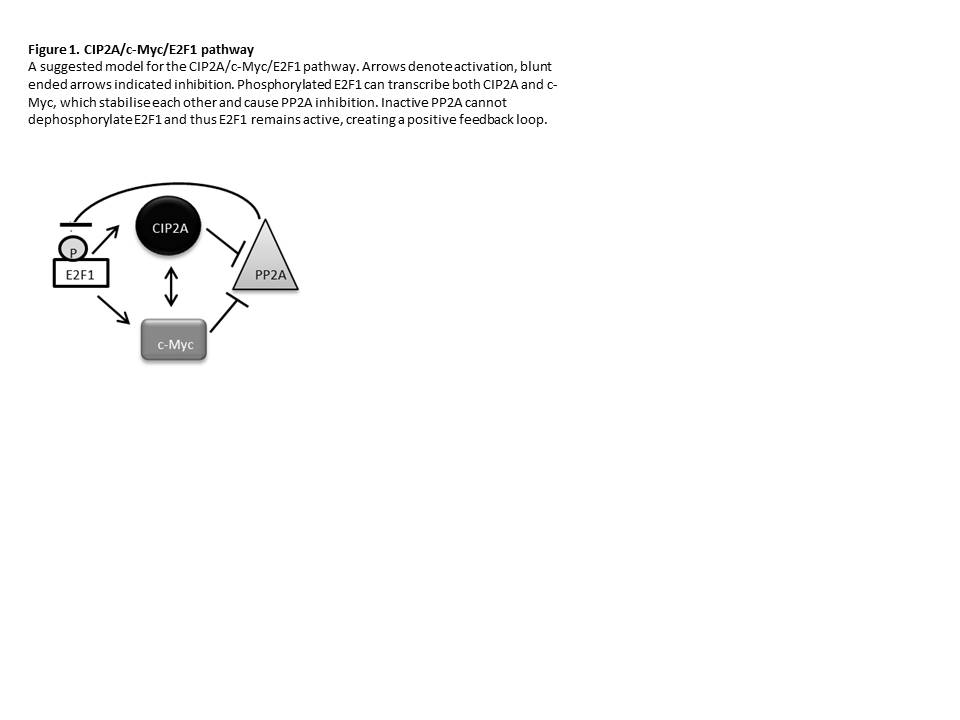
Our data demonstrate that E2F1 is stabilised in CML as a result of PP2A inactivation. CIP2A acts to inhibit PP2A leading to stabilisation of E2F1, creating a positive feedback loop generating constant transcription and stabilisation of both CIP2A and c-Myc (Figure 1). The positive feedback loop was interrogated by sequentially depleting CIP2A, c-Myc and E2F1 in K562 and CD34+ CML cells. Depleting CIP2A in CD34+ CML cells reactivates PP2A, resulting in the dephosphorylation of E2F1, and thus a decrease in E2F1 and c-Myc. This may occur by two mechanisms; firstly the decrease in CIP2A level means that CIP2A can no longer stabilise c-Myc, and secondly the reduction in E2F1 removes its drive to transcribe c-Myc. In CD34+ cells, c-Myc knockdown resulted in a decrease in CIP2A (p=0.04) and E2F1 (p=0.04). This is consistent with our model, whereby decreased CIP2A will reactivate PP2A, leading to the dephosphorylation of E2F1 and therefore a decrease in E2F1. Finally E2F1 was depleted in CD34+ cells. This resulted in a decrease in CIP2A (p=0.002) and c-Myc.
Summary
In summary, we have shown that a positive feedback loop occurs between CIP2A, E2F1 and c-Myc which inhibits PP2A activity and that only the 2G TKIs and not imatinib are able to supress it. Our study highlights a role for E2F1 in the CIP2A pathway, although it cannot be assumed that E2F1 is the only transcription factor regulating CIP2A and others may influence this dynamic process. Thus, CIP2A remains an attractive therapeutic target since high levels are only found in malignant cells.
Keyword(s): CML blast crisis, MYC, PP2A
Session topic: Novel actors in chronic myeloid leukemia biology
Type: Oral Presentation
Presentation during EHA20: From 13.06.2015 11:30 to 13.06.2015 11:45
Location: Room Stolz 1
Background
Impairment of PP2A activity by its negative regulators SET and cancerous inhibitor of PP2A (CIP2A) plays an important role in the pathogenesis and progression of chronic myeloid leukaemia (CML). CIP2A is associated with increased proliferation in several human malignancies and its over-expression can cause cellular transformation. High levels are an adverse prognostic indicator in many malignancies. In CML a high CIP2A protein level at diagnosis in imatinib treated patients may be a biomarker for subsequent blast crisis. However, its role in CML patients receiving second generation tyrosine kinase inhibitors (2G TKIs) is not known.
Aims
Our aim was to investigate if the superior clinical outcome seen in patients treated with 2G TKIs could be explained by modulation of the CIP2A pathway.
Methods
CIP2A, PP2A, PP2AY307, c-Myc and E2F1 proteins were assessed by FACS. CIP2A, c-Myc and E2F1 were depleted in K562 and CD34+ cells using siRNA.
Results
In mononuclear cells taken at diagnosis from 20 chronic phase CML patients, those with high CIP2A levels significantly decreased following dasatinib or nilotinib treatment (p=0.007 and p=0.001 respectively) and this was accompanied by a significant decrease in c-Myc, (p=0.002). Y307 phosphorylated (inactive) PP2A also decreased in dasatinib (p=0.03) and nilotinib (p=0.04) treated cells, thus increasing PP2A activity, but imatinib had no significant effect. These differences in TKI effects are not mediated by differential suppression of BCR-ABL activity (as assessed using the pCrKL assay).
E2F1 transcriptionally regulates both c-Myc and CIP2A and has previously been shown to be over-expressed in CML. E2F1 is high in patients with high diagnostic CIP2A protein levels compared to those with low CIP2A protein levels (p=0.04). Following 1 month of clinical 2G TKI treatment, E2F1 protein levels significantly decreased (p=0.01) and this was accompanied by a decrease in mRNA levels of its transcriptional target CIP2A. In sharp contrast, imatinib did not suppress E2F1, and thus CIP2A mRNA levels remain unchanged.
Our data demonstrate that E2F1 is stabilised in CML as a result of PP2A inactivation. CIP2A acts to inhibit PP2A leading to stabilisation of E2F1, creating a positive feedback loop generating constant transcription and stabilisation of both CIP2A and c-Myc (Figure 1). The positive feedback loop was interrogated by sequentially depleting CIP2A, c-Myc and E2F1 in K562 and CD34+ CML cells. Depleting CIP2A in CD34+ CML cells reactivates PP2A, resulting in the dephosphorylation of E2F1, and thus a decrease in E2F1 and c-Myc. This may occur by two mechanisms; firstly the decrease in CIP2A level means that CIP2A can no longer stabilise c-Myc, and secondly the reduction in E2F1 removes its drive to transcribe c-Myc. In CD34+ cells, c-Myc knockdown resulted in a decrease in CIP2A (p=0.04) and E2F1 (p=0.04). This is consistent with our model, whereby decreased CIP2A will reactivate PP2A, leading to the dephosphorylation of E2F1 and therefore a decrease in E2F1. Finally E2F1 was depleted in CD34+ cells. This resulted in a decrease in CIP2A (p=0.002) and c-Myc.
Summary
In summary, we have shown that a positive feedback loop occurs between CIP2A, E2F1 and c-Myc which inhibits PP2A activity and that only the 2G TKIs and not imatinib are able to supress it. Our study highlights a role for E2F1 in the CIP2A pathway, although it cannot be assumed that E2F1 is the only transcription factor regulating CIP2A and others may influence this dynamic process. Thus, CIP2A remains an attractive therapeutic target since high levels are only found in malignant cells.
Keyword(s): CML blast crisis, MYC, PP2A
Session topic: Novel actors in chronic myeloid leukemia biology