
Medicine

Contributions
Type: Oral Presentation
Presentation during EHA20: From 12.06.2015 11:30 to 12.06.2015 11:45
Location: Room Stolz 1
Background
Advanced malignancy often correlates with activation of the coagulation system, termed cancer coagulopathy, which is associated with increased mortality rates. Thrombin, a blood-derived serine protease, is the main effector of the coagulation cascade. There is convincing evidence that thrombin, by PAR-1 receptor, regulates numerous critical cellular events, including cell proliferation, cell adhesion, angiogenesis, and invasion. Dabigatran is a selective direct thrombin inhibitor that reversibly binds to thrombin. Dabigatran-bound thrombin is unable to cleave and activate PAR-1.
Aims
The purpose of this study is to explore if dabigatran may affect mechanisms favouring tumor growth by interfering with thrombin-induced PAR-1 activation.
Methods
The U87 glioblastoma cell line, the MCF-7 breast adenocarcinoma cell line and the endothelial cell line HUVEC, all expressing PAR-1, have been used for our experiments. Cell proliferation was measured by a colorimetric assay suitable for determining the number of viable cells. Cell death was quantitated by FACS analysis with PI/annexin. For RNA analysis, Real Time PCR was performed on a ABI PRISM 2600 instrument using SYBR Green. Protein expression was measured by WB analysis. Chemotaxis was studied using transwell plates. Endothelial cord formation was evaluated on Matrigel by enumerating branching points (tube formation) with an inverted microscope.
Results
Exposure of tumor cells to thrombin significantly increased cell proliferation as measured by MTS conversion. Using this approach, dabigatran was effective in antagonizing thrombin-induced proliferation in a dose-dependent manner. In our experimental system, U87 cells underwent cell death when cultured in starving medium. As thrombin has been shown to activate protein kinases interfering with caspases-dependent cell death, we thought to evaluate this property in U87 starved cultures.
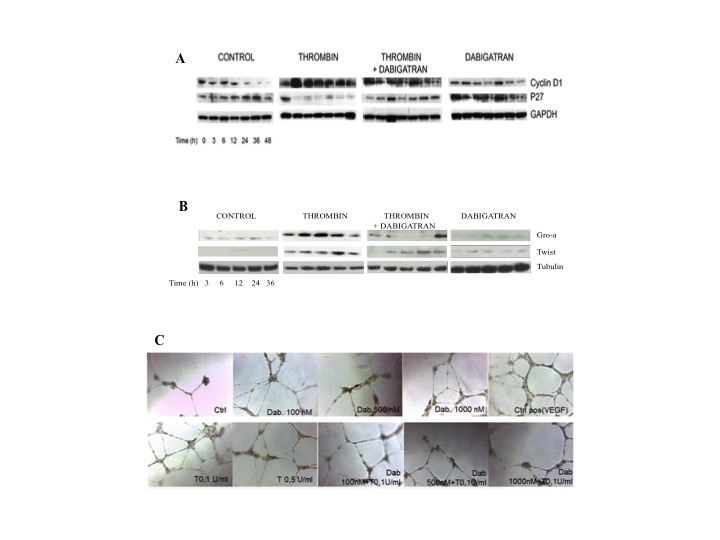
We found that the fraction of double positive annexin/PI cells was significantly lower in presence of thrombin and this protection was lost when dabigatran was coincubated with thrombin. We then evaluated the regulation of cell cycle proteins by thrombin and dabigatran. In particular, we evaluated the expression of cyclin D1 (promoting cell cycling) and p27 (inhibiting cell cycling) in U87 and MCF-7 starved cultures. Thrombin led to down-regulation of p27 with concomitant induction of cyclin D1. When dabigatran was added to cultures, the pattern of inhibition of growth observed in presence of starving medium was restored (fig.1 A). Treatment of MCF-7 cells with thrombin determined a slight but significant increase of the fraction of cells in S phase. The effect of thombin on cell cycle was completely antagonized by dabigatran.
We then evaluated the expression of pro-angiogenetic proteins, Twist and GRO-a, in MCF-7 cells and HUVEC cells following exposure to thrombin and dabigatran. We found that thrombin was significantly effective in inducing up-regulation of Twist and GRO-a mRNA in MCF7 and HUVEC cell lines. Expression of Twist was brought down to control levels when dabigatran was added to colture. WB analysis confirmed RT-PCR results (fig.1 B).
We also found that the chemoattractant effect of thrombin on tumor cells was lost in presence of dabigatran, and this effect was directly related to dabigatran concentrations.
FInally, vascular tube formation in HUVEC cells was increased in presence of thrombin and the induction of tube formation was progressively lost with dabigatran (fig.1 C).
Summary
Our data support a role of thrombin in inducing the proliferation, migration and pro-angiogenetic effects of tumor cells in-vitro. Dabigatran has shown activity in antagonizing all these effects, thereby impairing tumor growth and progression. In-vivo models may help to understand the relevance of this pathway.
Keyword(s): Cancer, Thrombin inhibitor, Thrombosis
Session topic: Thrombosis and vascular biology
Type: Oral Presentation
Presentation during EHA20: From 12.06.2015 11:30 to 12.06.2015 11:45
Location: Room Stolz 1
Background
Advanced malignancy often correlates with activation of the coagulation system, termed cancer coagulopathy, which is associated with increased mortality rates. Thrombin, a blood-derived serine protease, is the main effector of the coagulation cascade. There is convincing evidence that thrombin, by PAR-1 receptor, regulates numerous critical cellular events, including cell proliferation, cell adhesion, angiogenesis, and invasion. Dabigatran is a selective direct thrombin inhibitor that reversibly binds to thrombin. Dabigatran-bound thrombin is unable to cleave and activate PAR-1.
Aims
The purpose of this study is to explore if dabigatran may affect mechanisms favouring tumor growth by interfering with thrombin-induced PAR-1 activation.
Methods
The U87 glioblastoma cell line, the MCF-7 breast adenocarcinoma cell line and the endothelial cell line HUVEC, all expressing PAR-1, have been used for our experiments. Cell proliferation was measured by a colorimetric assay suitable for determining the number of viable cells. Cell death was quantitated by FACS analysis with PI/annexin. For RNA analysis, Real Time PCR was performed on a ABI PRISM 2600 instrument using SYBR Green. Protein expression was measured by WB analysis. Chemotaxis was studied using transwell plates. Endothelial cord formation was evaluated on Matrigel by enumerating branching points (tube formation) with an inverted microscope.
Results
Exposure of tumor cells to thrombin significantly increased cell proliferation as measured by MTS conversion. Using this approach, dabigatran was effective in antagonizing thrombin-induced proliferation in a dose-dependent manner. In our experimental system, U87 cells underwent cell death when cultured in starving medium. As thrombin has been shown to activate protein kinases interfering with caspases-dependent cell death, we thought to evaluate this property in U87 starved cultures.
We found that the fraction of double positive annexin/PI cells was significantly lower in presence of thrombin and this protection was lost when dabigatran was coincubated with thrombin. We then evaluated the regulation of cell cycle proteins by thrombin and dabigatran. In particular, we evaluated the expression of cyclin D1 (promoting cell cycling) and p27 (inhibiting cell cycling) in U87 and MCF-7 starved cultures. Thrombin led to down-regulation of p27 with concomitant induction of cyclin D1. When dabigatran was added to cultures, the pattern of inhibition of growth observed in presence of starving medium was restored (fig.1 A). Treatment of MCF-7 cells with thrombin determined a slight but significant increase of the fraction of cells in S phase. The effect of thombin on cell cycle was completely antagonized by dabigatran.
We then evaluated the expression of pro-angiogenetic proteins, Twist and GRO-a, in MCF-7 cells and HUVEC cells following exposure to thrombin and dabigatran. We found that thrombin was significantly effective in inducing up-regulation of Twist and GRO-a mRNA in MCF7 and HUVEC cell lines. Expression of Twist was brought down to control levels when dabigatran was added to colture. WB analysis confirmed RT-PCR results (fig.1 B).
We also found that the chemoattractant effect of thrombin on tumor cells was lost in presence of dabigatran, and this effect was directly related to dabigatran concentrations.
FInally, vascular tube formation in HUVEC cells was increased in presence of thrombin and the induction of tube formation was progressively lost with dabigatran (fig.1 C).
Summary
Our data support a role of thrombin in inducing the proliferation, migration and pro-angiogenetic effects of tumor cells in-vitro. Dabigatran has shown activity in antagonizing all these effects, thereby impairing tumor growth and progression. In-vivo models may help to understand the relevance of this pathway.
Keyword(s): Cancer, Thrombin inhibitor, Thrombosis
Session topic: Thrombosis and vascular biology